Doença de Wilson
Revisado por Dr Doug McKechnie, MRCGPÚltima atualização por Dr Philippa Vincent, MRCGPÚltima atualização 20 Jan 2025
Atende aos diretrizes editoriais
- BaixarBaixar
- Compartilhar
- Language
- Discussão
- Versão em Áudio
- Adicionar às fontes preferidas no Google
Nesta série:Testes de função hepáticaSíndrome de GilbertIcteríciaCirroseInsuficiência hepáticaColangite biliar primária
A doença de Wilson é um distúrbio genético em que o cobre se acumula no corpo, principalmente no fígado e no cérebro. Sem tratamento, o acúmulo de cobre pode causar sintomas graves. O tratamento é feito com medicação para remover o excesso de cobre e/ou para prevenir um acúmulo adicional de cobre.
Em resumo
Wilson's disease is a rare inherited condition where too much copper builds up in the body.
It is caused by a faulty gene that prevents the body from getting rid of excess copper.
Copper mainly builds up in the liver, brain, and the front layer of the eye.
Symptoms typically appear between ages 6 and 20, affecting the liver and brain.
Diagnosis involves blood and urine tests, eye examination, or a liver biopsy.
Treatment includes medicines to remove copper or block its absorption, and is lifelong.
Untreated Wilson's disease can be fatal, usually before age 40.
O que é a doença de Wilson?
A doença de Wilson é uma condição em que há um acúmulo excessivo de cobre no corpo. É um distúrbio hereditário raro que afeta cerca de 1 em cada 30.000 pessoas. Recebeu o nome do Dr. Samuel Wilson, que descreveu o distúrbio pela primeira vez em 1912.
Pessoas que herdam a falha genética na doença de Wilson não conseguem eliminar o cobre do corpo. O cobre é um metal traço presente em muitos alimentos. Pequenas quantidades de cobre são necessárias para manter a saúde. Normalmente, o corpo elimina qualquer excesso de cobre. Pessoas com a doença de Wilson não conseguem eliminar esse excesso de cobre, que se acumula no corpo, principalmente no fígado, no cérebro, na camada frontal do olho (chamada de córnea) e nos rins.
Excesso de cobre nas células do fígado (os hepatócitos) é prejudicial e leva a danos hepáticos. Danos ao tecido cerebral ocorrem principalmente em uma área chamada núcleo lenticular. Portanto, a doença de Wilson é às vezes também chamada de degeneração hepatolenticular.
O que causa a doença de Wilson?
Na doença de Wilson, um gene específico no cromossomo 13 não funciona. O gene é chamado ATP7B. Este gene normalmente controla a maneira como as células do fígado se livram do excesso de cobre. Normalmente, as células do fígado eliminam o excesso de cobre na bile. Se este processo não funcionar, o cobre se acumula nas células do fígado. Quando a capacidade de armazenamento de cobre das células do fígado é esgotada, o cobre se espalha na corrente sanguínea e deposita cobre em outras partes do corpo, principalmente no cérebro.
Como a doença de Wilson é herdada?
A doença de Wilson é um distúrbio autossômico recessivo. Isso significa que, para desenvolver a doença de Wilson, dois genes ATP7B anormais devem ser herdados - um de cada pai.
Herança de Wilson
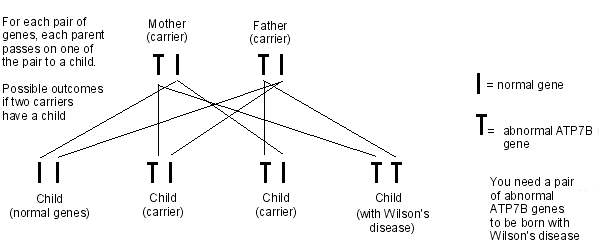
Pessoas que herdam apenas uma cópia do gene anormal são chamadas de portadoras. As portadoras não têm o distúrbio, pois possuem um gene normal que é suficiente para controlar a função do cobre no corpo. No entanto, as portadoras podem passar o gene anormal para seus filhos.
Quão comum é a doença de Wilson?
Cerca de 1 em cada 100 pessoas são portadoras do gene ATP7B. Quando duas pessoas que carregam o gene anormal têm um filho, há um:
1 em 4 chance de que a criança tenha a doença de Wilson (ao herdar o gene ATP7B anormal de ambos os pais).
2 em 4 chances de que a criança não tenha a doença de Wilson, mas seja portadora (herdando o gene ATP7B anormal de um dos pais, mas o gene normal do outro pai).
1 in 4 chance that the child will not have Wilson's disease, and will not be a carrier (by inheriting the normal gene from both parents).
Sintomas da doença de Wilson
Embora o defeito genético esteja presente desde o nascimento, leva vários anos para que o cobre se acumule a um nível em que comece a causar danos. Os sintomas geralmente começam a se desenvolver entre as idades de 6 e 20 anos, mais comumente na adolescência, mas algumas pessoas só começam a desenvolver sintomas na meia-idade.
Problemas no fígado
Os sintomas de problemas no fígado geralmente se desenvolvem primeiro. O efeito tóxico nas células do fígado pode causar inflamação do fígado (hepatite), o que pode causar:
Amarelamento da pele ou do branco dos olhos (icterícia).
Dor na barriga (abdominal).
Episódios de estar doente (vômito).
If left untreated, damage to liver cells causes scarring of the liver (cirrhosis). Eventually, cirrose grave and liver failure develop in untreated cases, causing severe problems.
(Nota: há várias causas de cirrose. A doença de Wilson é uma causa muito rara de cirrose.)
Problemas cerebrais
À medida que o cobre se deposita no cérebro, pode causar vários sintomas.
Physical symptoms, incluindo:
Tremor nos braços.
Lentidão de movimento.
Dificuldade na fala.
Problemas de escrita.
Dificuldade para engolir.
Uma caminhada instável.
Dores de cabeça.
Convulsões (ataques).
Sintomas psicológicos, incluindo:
Depressão.
Oscilações de humor
Incapacidade de se concentrar.
Uma mudança de personalidade levando a um comportamento argumentativo e emocional.
Severe problems. Se não tratado, o acúmulo de cobre no cérebro pode levar a:
Fraqueza muscular severa.
Rigidez severa.
Demência.
Olhos na doença de Wilson
Cobre pode se acumular na camada na frente do olho (chamada de córnea). Isso causa uma característica chamada anéis de Kayser-Fleischer - uma pigmentação amarronzada da córnea.
Anel de Kayser-Fleischer
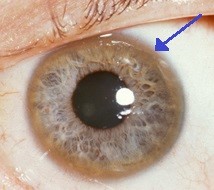
© Herbert L et al, CC BT 3.0, via Wikimedia Commons
Outras características da doença de Wilson
Outras características que podem se desenvolver incluem:
Anemia.
Dano renal.
Problemas cardíacos.
Inflamação do pâncreas (pancreatite).
Problemas menstruais.
Abortos espontâneos recorrentes em mulheres.
'Afinamento' prematuro dos ossos (osteoporose).
Como é diagnosticada a doença de Wilson?
Se houver suspeita de doença de Wilson, ela pode ser diagnosticada por meio de vários testes:
Um exame de sangue to measure caeruloplasmin. This is a protein that binds copper in the bloodstream. The level is low in nearly all people with Wilson's disease.
Outros exames de sangue também podem ser realizados. Estes podem ser feitos para medir os níveis de cobre e testar a função dos rins e do fígado.
Um especialista pode solicitar um exame de urina para medir a quantidade de cobre na urina ao longo de um período de 24 horas. A quantidade é tipicamente mais alta do que o normal.
Um exame da camada na frente do olho (chamada de córnea) por um óptico (optometrista) ou um especialista em olhos pode mostrar os anéis de Kayser-Fleischer se eles tiverem se desenvolvido. (Eles não estão presentes em todos os casos.)
A small sample (biopsy) of the liver may be taken to look at under the microscope. This can show the excess copper in the liver and the extent of any scarring of the liver (cirrhosis). Consulte o folheto separado chamado Biópsia do fígado para mais detalhes.
Other tests may also be advised - for example, a exame de ressonância magnética (RM).
Se a doença de Wilson for confirmada, os irmãos devem ser verificados para ver se também têm a condição. Irmãos e irmãs de uma pessoa com a doença de Wilson têm 1 em 4 chances de também ter a condição.
Tratamento da doença de Wilson
É essencial tratar a doença de Wilson, pois, sem tratamento, ela pode ser fatal. Quanto mais cedo o tratamento for iniciado, maior a chance de prevenir danos permanentes a longo prazo no fígado ou no cérebro. Não há cura para a doença de Wilson, mas o tratamento reduz os riscos de complicações.
Penicilamina is a medicine called a chelating agent and is used to remove copper from the body. The penicillamine causes the excess copper from the body to be passed out in the urine. The dose may be reduced to a maintenance dose after about a year when the initial build-up of copper has been cleared. Unfortunately, the symptoms of Wilson's disease can deteriorate as a side effect of the medication.
Trientina is an alternative to penicillamine. It too is a chelating agent and removes copper from the body. It has slightly fewer side effects.
Zinco is an option in certain circumstances. Zinc works by blocking the gut from absorbing copper from food. Therefore, it does not clear excess copper from the body, but prevents any further build-up of copper. Zinc is much less likely than penicillamine or trientine to cause side-effects. It may be an option for people who are diagnosed at the very early stages of the disease and have no symptoms. Also, a switch to zinc may be an option for people who have been initially treated with penicillamine or trientine once the initial build-up of copper has been cleared from the body. Zinc may also be taken by pregnant women.
Nota: o tratamento é necessário para a vida. Primeiro, é usado para eliminar o excesso de cobre e depois para prevenir o acúmulo futuro de cobre. A falha em tomar a medicação pode levar ao retorno do acúmulo de cobre, o que pode ser grave - até mesmo fatal.
Para as poucas pessoas que não respondem ao tratamento com medicação, ou são diagnosticadas em estágio avançado da doença com cicatrização severa do fígado (cirrose) ou insuficiência hepática, um transplante de fígado pode ser uma opção. Isso pode salvar vidas. A perspectiva a longo prazo após um transplante de fígado é geralmente muito boa.
Dieta
Alimentos com alta concentração de cobre geralmente devem ser evitados, pelo menos no primeiro ano de tratamento, quando o excesso de cobre está sendo eliminado do corpo. Isso inclui fígado, chocolate, nozes, cogumelos e frutos do mar, especialmente lagosta.
Complicações da doença de Wilson
Se o tratamento for iniciado nos estágios iniciais da doença, geralmente funciona muito bem. Pode-se esperar uma duração e qualidade de vida normais.
No entanto, sem qualquer tratamento, a doença de Wilson é geralmente fatal - tipicamente antes dos 40 anos de idade.
If symptoms have developed before treatment has started, some of the symptoms improve with treatment but some may remain permanently. For example, some of the brain symptoms are permanent once they develop. A specialist will be able advise about which symptoms may go and which may be permanent, once treatment begins.
Escolhas do paciente para Condições genéticas

Saúde infantil
Deficiência de alfa-1 antitripsina
A deficiência de alfa-1 antitripsina é uma condição genética hereditária. Uma condição genética é aquela que pode ser transmitida dos pais através dos genes. Na deficiência de alfa-1 antitripsina, o resultado de uma anormalidade genética leva a danos nos pulmões e, em algumas pessoas, no fígado. Os sintomas pulmonares são os mais comuns e incluem falta de ar, tosse e chiado. Os sintomas podem piorar com o tempo. Atualmente, não há cura para a deficiência de alfa-1 antitripsina. O tratamento visa retardar a progressão da doença.
por Dra. Philippa Vincent, MRCGP

Saúde infantil
Malformação congênita das vias aéreas pulmonares
A malformação congênita das vias aéreas pulmonares (CPAM) é uma anomalia rara do desenvolvimento pulmonar. É encontrada em bebês não nascidos ou em bebês jovens. O nome foi recentemente alterado de malformação adenomatoide cística congênita (CCAM). Está sendo cada vez mais detectada pelo ultrassom de rotina durante a gravidez. Congênito significa presente desde o nascimento. Pulmonar significa relacionado aos pulmões. A gravidade da anomalia é muito variável. Algumas lesões podem encolher ou até desaparecer sem tratamento. Algumas lesões fazem com que o bebê tenha problemas respiratórios logo após o nascimento. Algumas lesões causam problemas graves para o bebê e podem ter um desfecho ruim.
por Dra. Hayley Willacy, FRCGP
Perguntas frequentes
If I am a carrier of the Wilson's disease gene, can I still pass it on even if I don't have the condition?
Yes, if you inherit only one copy of the abnormal gene, you are considered a carrier. Carriers do not develop Wilson's disease themselves because they have one normal gene that is sufficient to control copper function. However, carriers can pass the abnormal gene on to their children.
What is the likelihood of a child having Wilson's disease if both parents are carriers?
If both parents are carriers of the abnormal ATP7B gene, there is a 1 in 4 chance that their child will inherit two abnormal genes and therefore develop Wilson's disease. There is also a 2 in 4 chance the child will be a carrier and a 1 in 4 chance the child will not have Wilson's disease and not be a carrier.
What happens if treatment for Wilson's disease is not followed consistently?
Treatment for Wilson's disease is needed for life. Initially, it clears excess copper, and then it prevents future accumulation. Failure to take the medication can lead to a return of copper build-up, which can be serious and even fatal.
Which specific foods should be limited or avoided if I have Wilson's disease?
Foods with a high concentration of copper should generally be avoided, especially during the first year of treatment when excess copper is being cleared from the body. These include liver, chocolate, nuts, mushrooms, and shellfish, particularly lobster.
What is the long-term outlook for someone who has a liver transplant for Wilson's disease?
For the few people who do not respond to medication or are diagnosed late with severe liver damage or liver failure, a liver transplant can be a life-saving option. The long-term outlook after a liver transplant is usually very good.
Leitura adicional e referências
- British Liver Trust
- Doença de Wilson; Herança Mendeliana Online no Homem (OMIM)
- Diretrizes de Prática Clínica da EASL: Doença de Wilson. J Hepatol. Mar 2012;56(3):671-85. doi: 10.1016/j.jhep.2011.11.007.
- Fundação para Doenças Hepáticas Infantis
- Grupo de Apoio à Doença de Wilson do Reino Unido
- Compra R; O tratamento da doença de Wilson, um raro distúrbio genético do metabolismo do cobre. Sci Prog. 2013;96(Pt 1):19-32.
- Aggarwal A, Bhatt M; Atualização sobre a doença de Wilson. Int Rev Neurobiol. 2013;110:313-48. doi: 10.1016/B978-0-12-410502-7.00014-4.
- Bandmann O, Weiss KH, Kaler SG; Doença de Wilson e outros distúrbios neurológicos relacionados ao cobre. Lancet Neurol. 2015 Jan;14(1):103-13. doi: 10.1016/S1474-4422(14)70190-5.
Sobre o autorVer biografia completa

Dr Philippa Vincent, MRCGP
Médico Generalista, Autor Médico
MB BS, Bsc, MRCGP (2000), DCH, DFSRH, DRCOG
Dra Philippa Vincent é um médico do NHS trabalhando no norte de Londres.
Sobre o revisorVer biografia completa

Dr Doug McKechnie, MRCGP
Redator Médico
MA, MBBS, MSc, DRCOG, MRCP(UK), MRCGP(2021), FHEA
O Dr. Doug McKechnie é um médico do NHS que trabalha em Londres. Ele trabalha em tempo integral na prática clínica e também é o Vice-Líder do módulo de Prática Clínica e Profissional na Faculdade de Medicina da University College London.
Histórico do artigo
As informações nesta página são escritas e revisadas por clínicos qualificados.
Artigo também disponível em Inglês, Alemão, Espanhol, Francês, Italiano, Português, Hindi, Hebraico, Árabe, e Sueco.
Próxima revisão prevista: 19 Jan 2028
20 Jan 2025 | Última versão

Pergunte, compartilhe, conecte-se.
Navegue por discussões, faça perguntas e compartilhe experiências em centenas de tópicos de saúde.

Sentindo-se mal?
Avalie seus sintomas online gratuitamente
Inscreva-se no boletim informativo do Patient
Sua dose semanal de conselhos de saúde claros e confiáveis - escritos para ajudá-lo a se sentir informado, confiante e no controle.
Ao se inscrever, você aceita nossos Política de Privacidade. Você pode cancelar a inscrição a qualquer momento. Nunca vendemos seus dados.
Mais sobre saúde infantil
- Ansiedade em crianças
- Imunização BCG
- Doença cardíaca congênita
- Morte súbita infantil
- Depressão em crianças
- Diabetes em crianças
- Gastroenterite em crianças
- Vacina contra HPV
- Dor abdominal no lado esquerdo em crianças
- Vacina meningocócica para meningite
- Distrofia muscular
- Neuroblastoma
- Vulvovaginite pediátrica
- Estenose pilórica
- Sangramento retal em crianças
- Vírus sincicial respiratório (RSV)
- Crescimento restrito
- Retinopatia da prematuridade
- Bloqueio do ducto lacrimal em bebês
- Tétano e a vacina contra o tétano