Doença de Huntington
Revisado por Dr Colin Tidy, MRCGPÚltima atualização por Dr Toni Hazell, MRCGPÚltima atualização 21 Dez 2023
Atende aos diretrizes editoriais
- BaixarBaixar
- Compartilhar
- Language
- Discussão
- Versão em Áudio
- Adicionar às fontes preferidas no Google
A doença de Huntington é uma condição geneticamente herdada que faz com que partes do cérebro e do sistema nervoso parem de funcionar corretamente ao longo do tempo. É uma condição de progressão lenta que interfere nos movimentos do seu corpo, no pensamento e no julgamento, e pode levar a uma mudança no seu comportamento.
Os sintomas ocorrem devido a danos e morte de algumas células cerebrais (neurônios) em partes específicas do seu cérebro. Testes genéticos ajudam a diagnosticar a doença de Huntington.
Atualmente, não há cura para a doença de Huntington. O tratamento visa tentar controlar os sintomas o máximo possível quando eles se desenvolvem.
Em resumo
Huntington's disease is a genetic condition that damages brain function and worsens over time.
It can cause problems with movement, thinking, judgement, and behaviour.
Symptoms often appear between the ages of 30 and 50 but can occur earlier.
There is no cure, but treatments help manage the symptoms.
If you have neurological symptoms, especially with a family history of Huntington's disease, see a doctor.
O que é a doença de Huntington?
A doença de Huntington é uma condição geneticamente herdada que danifica gradualmente a função cerebral. Ela progride ao longo do tempo, muitas vezes se tornando fatal entre os 40 e 50 anos de idade.
Isso pode levar a uma mudança no seu comportamento e interferir em:
Movimentos do seu corpo.
Raciocínio.
Consciência.
Pensamento.
Julgamento (cognição).
A doença foi nomeada em homenagem a George Huntington, que a descreveu pela primeira vez em 1872.
Quão comum é a doença de Huntington?
A doença de Huntington afeta entre 11 - 14 pessoas por 100.000 no Reino Unido, no resto da Europa e na América do Norte. Em todo o mundo, parece ser mais comum entre populações brancas do que entre pessoas asiáticas ou africanas. A doença de Huntington afeta homens e mulheres igualmente.
Quão comum é a doença de Huntington juvenil?
Entre 5 a 10 em cada 100 pessoas com a doença de Huntington desenvolvem sintomas antes dos 20 anos. Isso é conhecido como doença de Huntington juvenil.
Isso pode causar sintomas mais graves. A maioria das pessoas que tem esse tipo de doença de Huntington herda a condição do pai.
Quais são os sintomas da doença de Huntington?
Os sintomas da doença de Huntington podem variar de pessoa para pessoa e piorar gradualmente ao longo do tempo. Os sintomas podem se desenvolver a qualquer momento, mas tendem a surgir entre os 30 e 50 anos de idade.
Os sintomas são agrupados em três áreas:
Movimento.
Cognitivo.
Problemas de humor e comportamento.
Problemas de movimento
A doença de Huntington pode causar problemas de movimento tanto involuntários quanto prejudicados, que incluem:
Movimentos bruscos e involuntários na cabeça, rosto, braços ou pernas (coreia).
Espasmos musculares principalmente nos ombros, pescoço, braços e pernas (distonia).
Membros rígidos ou enrijecidos e movimentos mais lentos (bradicinesia).
Dificuldade para engolir.
Fala arrastada.
Movimentos oculares lentos.
Na coreia grave, você pode desenvolver movimentos incontroláveis dos braços ou pernas (chamados balismo). Isso pode interferir na sua capacidade de se movimentar. Você pode ter mais probabilidade de cair, ter dificuldade em segurar objetos e em se alimentar ou vestir.
À medida que a doença de Huntington progride, os sintomas de coreia tendem a ser gradualmente substituídos por distonia. A distonia pode levar a movimentos de torção, movimentos repetitivos ou posturas anormais.
Porque a doença também pode afetar os músculos que controlam a deglutição, engasgos podem ser um problema, assim como a perda de peso devido à dificuldade em comer.
Os movimentos oculares lentos e incomuns podem dificultar olhar de um lado para o outro ou para cima e para baixo.
Problemas cognitivos
Como a doença de Huntington afeta tanto o cérebro quanto o sistema nervoso, podem surgir problemas cognitivos, incluindo:
Perda de memória de curto prazo e lapsos de memória.
Falta de concentração; não conseguir focar, organizar ou priorizar tarefas.
Dificuldade em processar pensamentos.
Problemas de orientação.
Dificuldade em aprender novas habilidades ou informações.
Julgamento prejudicado.
Falta de controle de impulso.
O declínio lento na cognição pode ser semelhante a um problema do tipo demência.
Problemas de humor e comportamento
Mudanças no seu comportamento podem ser um dos primeiros sinais da doença de Huntington e podem surgir antes de seus movimentos serem afetados. Isso inclui:
Irritabilidade, apatia ou tristeza.
Baixo interesse em autocuidado; por exemplo, lavar-se com menos frequência, não cuidar da aparência e estar desarrumado.
Mental health problems including depressão or suicidal thoughts.
A depressão é o sintoma psicológico mais comum em pessoas com a doença de Huntington e há um risco aumentado de suicídio.
Outros problemas de saúde mental incluem:
As mudanças de personalidade relacionadas à doença de Huntington podem ser muito difíceis para você ou seus entes queridos lidarem, e podem colocar uma pressão sobre seus relacionamentos.
Você pode achar difícil aceitar que seu comportamento pode ser um problema. Isso faz parte da maneira como a doença de Huntington afeta seu cérebro
Sintomas da doença de Huntington juvenil
A doença de Huntington juvenil é menos comum do que a forma que se manifesta mais tarde. Os sintomas começam na infância ou adolescência (antes dos 20 anos). Cerca de 5 - 10% de todos os casos de doença de Huntington se manifestam nessa idade. Os sintomas são semelhantes aos da doença de Huntington que se manifesta em adultos, apenas ocorrem mais cedo.
Quando consultar um médico
Se você estiver experimentando qualquer tipo de sintoma neurológico, é importante consultar um médico - há uma ampla variedade de possíveis causas, incluindo a doença de Huntington e outras causas. Se você sabe que tem um histórico familiar de doença de Huntington, certifique-se de mencionar isso na consulta.
O que causa a doença de Huntington?
A doença de Huntington é causada por um gene defeituoso que você herda dos seus pais.
A doença de Huntington é uma condição autossômica dominante, o que significa que você pode herdar a doença de apenas um dos seus pais.
Se um dos seus pais tem uma cópia defeituosa do gene, há uma chance de 50:50 de que cada filho que eles tiverem herde o gene defeituoso.
Se um membro da sua família que carrega o gene defeituoso morrer antes de seus sintomas se desenvolverem (e, portanto, antes que a doença de Huntington seja diagnosticada), os parentes não estarão cientes do histórico familiar.
Como a doença de Huntington afeta o cérebro
O defeito genético na doença de Huntington significa que certas proteínas necessárias para produzir substâncias químicas cerebrais não podem ser produzidas no seu cérebro como normalmente. Acredita-se que isso leva a danos e morte de algumas células cerebrais em partes do seu cérebro chamadas gânglios basais e córtex.
É esse dano que leva aos sintomas da doença de Huntington. Há também um acúmulo de uma substância química chamada dopamina no cérebro, que contribui para os problemas de movimento.
Novas mutações
Ocasionalmente, alguém com a doença de Huntington pode não ter um histórico da doença na família. Isso pode ocorrer devido ao que é chamado de 'nova mutação'.
Uma nova mutação é uma falha em um gene que está presente pela primeira vez em um membro da família. Isso pode acontecer devido a:
Uma falha no material genético, seja no óvulo ou no esperma de um dos pais da pessoa afetada.
Uma falha no material genético do embrião.
Não está claro o que causa essa mutação.
Como é diagnosticada a doença de Huntington?
Testes genéticos is used to confirm whether you carry the faulty gene that causes Huntington's disease.
É recomendado que você passe por aconselhamento se estiver considerando fazer testes genéticos. Seu médico poderá encaminhá-lo a um especialista que é um conselheiro genético.
Outros exames
tomografia computadorizada ou ressonância magnética of your brain may show some typical signs of Huntington's disease. However, these scans are not usually helpful in the early stages as changes may not be present. Scanning is mostly used in the later stages of the disease.
Qual é o tratamento para a doença de Huntington?
Atualmente, não há cura para a doença de Huntington. Além disso, nenhum tratamento foi encontrado para atrasar o início ou a progressão dos sintomas. Portanto, o tratamento visa tentar controlar os sintomas o máximo possível quando eles se desenvolvem. Estes incluem:
Medicamentos para problemas físicos.
Medicamentos para problemas psicológicos.
Fisioterapia.
Terapia ocupacional.
Terapia da fala.
Orientação nutricional.
Aconselhamento, orientação e apoio
Medicamentos para problemas físicos
Existem vários medicamentos diferentes que podem ser usados para ajudar a tratar problemas físicos. Estes incluem:
Benzodiazepines, for example, clonazepam e diazepam para controlar a coreia.
Tetrabenazine também pode ser usado.
Agonistas da dopamina e levodopa para gerenciar a bradicinesia.
Decidir se e quando iniciar o tratamento dependerá de encontrar um equilíbrio entre os benefícios e os efeitos colaterais.
Medicamentos para problemas psicológicos
Se você desenvolver depressão, medicamentos antidepressivos podem ser úteis. Medicamentos também estão disponíveis para tratar alguns outros problemas de saúde mental que podem estar associados à doença de Huntington.
Fisioterapia
Você pode ser encaminhado a um fisioterapeuta para obter ajuda com exercícios para o seu equilíbrio e exercícios para ajudá-lo a se movimentar com mais facilidade.
Terapia ocupacional
Um terapeuta ocupacional pode ajudá-lo com quaisquer adaptações que você precise para tornar sua vida diária mais fácil. Por exemplo, eles podem ajudar com adaptações em sua casa, como acesso para cadeiras de rodas, corrimãos e mudanças no seu quarto e banheiro.
Terapia da fala
Um terapeuta de fala e linguagem pode ajudar com dificuldades de fala e/ou deglutição. Eles podem ensinar diferentes formas de comunicação.
Conselhos dietéticos
Você pode ser encaminhado a um nutricionista se perder muito peso (devido a dificuldades de deglutição e problemas de movimento). Eles podem aconselhar sobre alimentos que podem ser mais fáceis para você comer porque exigem menos mastigação.
Às vezes, problemas de deglutição significam que pode ser necessário um tubo nasogástrico. Este é um tubo que passa pelo seu nariz até o estômago, permitindo que o alimento seja entregue ao seu estômago sem que você precise engolir.
Outros tratamentos
A maioria das pessoas com doença de Huntington tem uma equipe de especialistas em saúde que trabalham com elas. Seu médico de família geralmente coordena seu cuidado. Outros membros da equipe podem incluir um especialista em problemas cerebrais, nervosos e musculares (um neurologista), um psiquiatra e um conselheiro genético.
Possíveis tratamentos futuros
Vários novos tratamentos para a doença de Huntington estão sendo estudados. Eles incluem tratamentos de terapia genética e vários medicamentos. Por exemplo, estão em andamento ensaios que analisam o efeito de um novo medicamento chamado pridopidina.
Os ensaios clínicos também estão investigando medicamentos para prevenir que pessoas com o gene defeituoso da doença de Huntington desenvolvam a doença. No entanto, esses tratamentos ainda estão em um nível muito experimental.
Mais pesquisas são necessárias antes de sabermos se tais tratamentos são úteis para a doença de Huntington.
Qual é a perspectiva para a doença de Huntington?
A doença de Huntington progride lentamente e os sintomas aumentam e pioram com o tempo. Nos estágios mais avançados da doença de Huntington, você se tornará totalmente dependente de outras pessoas e precisará de cuidados em tempo integral. A doença de Huntington leva a uma incapacidade considerável e, atualmente, eventualmente levará à morte.
Atualmente, a maioria das pessoas vive entre 10 a 25 anos após desenvolverem os primeiros sintomas da doença de Huntington. Alguém com a doença de Huntington geralmente morre de uma infecção, como pneumonia, mas o suicídio é muito comum.
Remember that new treatments are under investigation. Your specialist and the Associação de Doença de Huntington will be able to discuss any treatment developments or trial treatments with you.
Posso transmitir a doença de Huntington para meus filhos?
Se você carrega o gene defeituoso da doença de Huntington, para cada filho que você tiver, há uma chance de 50:50 de que eles também tenham a doença de Huntington.
If you or your partner have Doença de Huntington, prenatal testing is available. This can show whether your baby has the defective gene and therefore whether they will develop the doença. No entanto, os testes nem sempre são 100% precisos.
Herança autossômica dominante
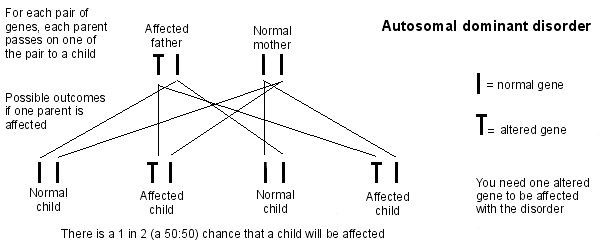
O diagnóstico genético pré-implantacional (DGP) também está disponível se um dos pais carregar o gene defeituoso. Isso basicamente envolve o casal se submeter a um tratamento do tipo FIV para que os embriões possam ser testados para a doença de Huntington antes de serem implantados no útero da mulher. Somente embriões sem o gene defeituoso da doença de Huntington são implantados.
Recomenda-se aconselhamento genético e orientação especializada se você ou seu parceiro tiverem a doença de Huntington, ou se houver casos de doença de Huntington em qualquer uma das suas famílias, e vocês estiverem considerando uma gravidez.
Escolhas do paciente para Distúrbios do movimento

Cérebro e nervos
Síndrome de Tourette
Tourette syndrome is a condition that causes you to make involuntary movements or noises called tics. It tends to be associated with various other problems such as behavioural problems and attention deficit hyperactivity disorder (ADHD). It can often be well managed with psychological treatments and sometimes with medication. Simple motor tics are brief and sudden repetitive movements that involve just a few muscle groups. Simple vocal tics involve saying a word or phrase, or making a different noise. Complex tics involve several different muscle groups in areas throughout your body. Complex motor tics include jumping, bending, twisting or other complex movements. Complex vocal tics include repeating other people’s words or phrases (echolalia), or using obscene, vulgar or swear words (coprolalia). The most common of all childhood tics is Tourette syndrome.
por Dr. Colin Tidy, MRCGP

Cérebro e nervos
Tremor essencial
Tremor essencial é o termo usado para descrever movimentos involuntários de tremor em partes do seu corpo - mais comumente nos braços e mãos.
por Dra. Hayley Willacy, FRCGP
Perguntas frequentes
What is the timeline of Huntington's disease from symptom onset to end of life?
Huntington's disease is a progressive condition where symptoms gradually increase and worsen over time. Most people live for 10 to 25 years after they first develop symptoms. In the later stages, individuals become fully dependent on others and require full-time care, eventually leading to death.
What is the average age when symptoms of Huntington's disease typically appear?
Symptoms of Huntington's disease can develop at any time, but they most commonly start between the ages of 30 and 50. In some cases, juvenile Huntington's disease can manifest before the age of 20.
When should I see a doctor if I suspect I might have Huntington's disease?
If you are experiencing any neurological symptoms, it's important to consult a doctor. There are many possible causes for such symptoms. If you have a family history of Huntington's disease, you should definitely mention this to your doctor during the appointment.
How does Huntington's disease progress from chorea to dystonia?
In the early stages of Huntington's disease, an individual might experience chorea, which involves jerky, involuntary movements. As the disease progresses, these chorea symptoms tend to be gradually replaced by dystonia, which can lead to twisting movements, repetitive movements, or abnormal postures.
Are new treatments for Huntington's disease being developed?
Yes, various new treatments for Huntington's disease are currently being studied. These include gene therapy treatments and different medicines. Clinical trials are also investigating medicines aimed at preventing people with the faulty gene from developing the disease. However, these treatments are still experimental, and more research is needed to determine their effectiveness.
What mental health conditions are associated with Huntington's disease?
People with Huntington's disease can experience a range of mental health problems. Depression is a very common psychological symptom, and there is an increased risk of suicide. Other associated conditions include obsessive-compulsive disorder, bipolar disorder, and schizophrenia.
If I have Huntington's disease, how can I ensure my future children won't inherit it?
If you or your partner carry the defective gene, prenatal testing is available to determine if your baby has the gene. Another option is Preimplantation Genetic Diagnosis (PGD), which involves using IVF-like treatment to test embryos for the defective gene before implantation. Only embryos without the faulty gene are implanted. Genetic counselling is recommended to discuss these options and provide specialist advice.
Leitura adicional e referências
- Roos RA; Doença de Huntington: uma revisão clínica. Orphanet J Rare Dis. 20 de dezembro de 2010;5(1):40. doi: 10.1186/1750-1172-5-40.
- Zielonka D, Mielcarek M, Landwehrmeyer GB; Atualização sobre a doença de Huntington: avanços nos cuidados e opções terapêuticas emergentes. Parkinsonism Relat Disord. 2015 Mar;21(3):169-78. doi: 10.1016/j.parkreldis.2014.12.013. Epub 2014 Dec 19.
- Rede Europeia de Doença de Huntington; Informações sobre o projeto EHDN e estudos participantes
- Pandey M, Rajamma U; Doença de Huntington: a chegada da maturidade. J Genet. 2018 Jul;97(3):649-664.
- Rawlins MD, Wexler NS, Wexler AR, et al; A Prevalência da Doença de Huntington. Neuroepidemiologia. 2016;46(2):144-53. doi: 10.1159/000443738. Publicado online em 30 de janeiro de 2016.
- Caron NS, Wright GEB, Hayden MR; Doença de Huntington GeneReviews® 23 de outubro de 1998 [atualizado em 11 de junho de 2020].
- Marcus R; O que é a Doença de Huntington? JAMA. 12 de setembro de 2023;330(10):1014. doi: 10.1001/jama.2023.13024.
Sobre o autorVer biografia completa

Dra. Toni Hazell, MRCGP
MBBS, BSc, MRCGP, DFSRH, Dip GU med, DRCOG, DCH (London, UK, 2000)
A Dra. Toni Hazell se formou na Escola de Medicina do Hospital St. Mary e fez seu VTS no Hospital Northwick Park.
Sobre o revisorVer biografia completa

Dr Colin Tidy, MRCGP
Médico Generalista, Autor Médico
MBBS, MRCGP, MRCP (Paediatrics), DCH
Dr Colin Tidy é um médico do NHS, baseado em Oxfordshire.
Histórico do artigo
As informações nesta página são escritas e revisadas por clínicos qualificados.
Artigo também disponível em Inglês, Alemão, Espanhol, Francês, Italiano, Português, Hindi, Hebraico, Árabe, e Sueco.
Próxima revisão prevista para: 19 Dez 2028
21 Dez 2023 | Última versão

Pergunte, compartilhe, conecte-se.
Navegue por discussões, faça perguntas e compartilhe experiências em centenas de tópicos de saúde.

Sentindo-se mal?
Avalie seus sintomas online gratuitamente
Inscreva-se no boletim informativo do Patient
Sua dose semanal de conselhos de saúde claros e confiáveis - escritos para ajudá-lo a se sentir informado, confiante e no controle.
Ao se inscrever, você aceita nossos Política de Privacidade. Você pode cancelar a inscrição a qualquer momento. Nunca vendemos seus dados.
Mais sobre cérebro e nervos
- síndrome do túnel do carpo
- Síndrome da cauda equina
- Cefaleias em salvas
- Epilepsia
- Medicação para epilepsia e efeitos colaterais
- Pé caído
- Transtorno neurológico funcional
- síndrome de Guillain-Barré
- Cefaleia por uso excessivo de medicação
- Enxaqueca e contracepção hormonal combinada
- tratamento da enxaqueca
- Neuralgia pós-herpética
- Mutismo seletivo
- Herpes zoster
- Acidente Vascular Cerebral (AVC)
- Morte súbita inesperada na epilepsia
- Cefaleia tensional
- Crises tônico-clônicas
- Ataque isquêmico transitório
- Neuralgia do trigêmeo